Page 1 of 2
Cutoff lengths in the Jastrow factor
Posted: Tue Jun 11, 2013 11:20 am
by Sweyn Asleifsson
Can anyone advise me on how to choose the cutoff lengths of u, chi and f in the Jastrow factor for graphene? They don't seem to change much when I optimise them, but if I fix them at different values and optimise the other parameters I get significantly different VMC energies.
Thanks,
S.
Re: Cutoff lengths in the Jastrow factor
Posted: Tue Jun 11, 2013 7:56 pm
by Neil Drummond
Dear Sweyn,
The usual advice is to restrict the cutoff lengths for chi and f to something of the order of a bond-length or so, but to allow the cutoff length for u to be as large as possible (ideally the radius of the largest sphere that can be inscribed in the WIgner-Seitz cell of the simulation cell). However, 2D systems are highly anisotropic and long-range isotropic 2-body terms can lead to instability in DMC. I suggest limiting the cutoff length for u to be less than, say, 10 a.u. This seems to ensure there are no problems in DMC.
Best wishes,
Neil.
Re: Cutoff lengths in the Jastrow factor
Posted: Fri Sep 06, 2013 10:47 am
by varelse
I did a variance-optimization VMC run for a benzene molecule. I took the starting value for L_chi as 9.3 which is about the "diameter" of the molecule, but after 9 steps of optimization it reduced to nearly 3 (i didn't use f term to reduce the number of parameters). Is this behaviour correct, or rather is this an effect of numerical instability? Maybe this is also the effect of two-dimensionality?
Re: Cutoff lengths in the Jastrow factor
Posted: Fri Sep 06, 2013 12:07 pm
by Mike Towler
Hi,
Neil said that one should start the cutoff length for chi to "something of the order of a bond-length or so".
The bond length in benzene is around 2.62 au.
You start off with 9.3 au, and it optimizes to ''nearly 3", which is indeed "of the order of a bond length or so".
So your optimization behaves exactly as Neil said it would, no?
Mike
Re: Cutoff lengths in the Jastrow factor
Posted: Fri Sep 06, 2013 9:08 pm
by varelse
Certainly, there is a typo in my question, I meant L_u not L_chi. L_chi started as "the order of bond length or so" and remained in such range.
Re: Cutoff lengths in the Jastrow factor
Posted: Sat Sep 07, 2013 8:32 pm
by Neil Drummond
Dear Varelse,
It is unusual for the cutoff length of u to shrink like that, although certainly not impossible. Did the energy & variance decrease over successive optimisation cycles?
Best wishes,
Neil.
Re: Cutoff lengths in the Jastrow factor
Posted: Mon Sep 09, 2013 11:41 am
by varelse
Yes. It decreases in about 5 optimization steps, and then oscillates within the error bar, like it should.
Now, I am trying different starting values.
For starting values 8 and 3 I get also the "final" L_u about 3, with "proper" energy an variance behaviour (first decrease, then oscillations around a constant value). For starting value 11 I got Lu 3.51 after ten optimization steps, but I am not sure if variance reached a stable value, and also L_u was constantly decreasing, so I would better continue this run. The starting values below 3 seem to have a bit weird behaviour in cutoff optimization, the L_u reaches a stable value, which remains the same for a few optimization steps, and then increases. They also seem to give higher energy and variances, but I have to give them more optimization steps to be sure that the energy and variance converges, and to see what happens with the cutoff.
So I suspect that 3 is a value to which the L_u optimization will always converge, but sometimes very slowly, and I need more calculation to see if this is true.
Re: Cutoff lengths in the Jastrow factor
Posted: Sat Oct 12, 2013 11:51 pm
by varelse
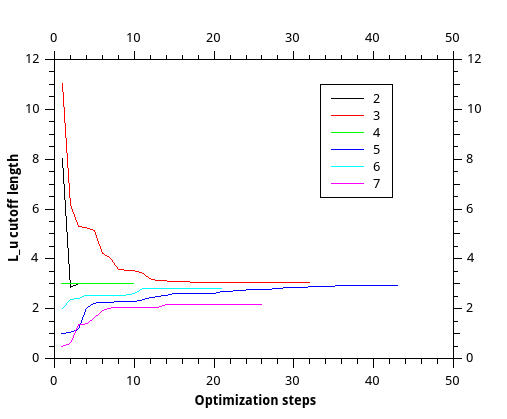
As for the benzene case, here is the
graph showing L_u evolution during the optimization, starting from different values, so probably the drop is not just the numerical instability, but the right behviour for this systems.
http://www.mediafire.com/convkey/481e/z ... z2xcfg.jpg
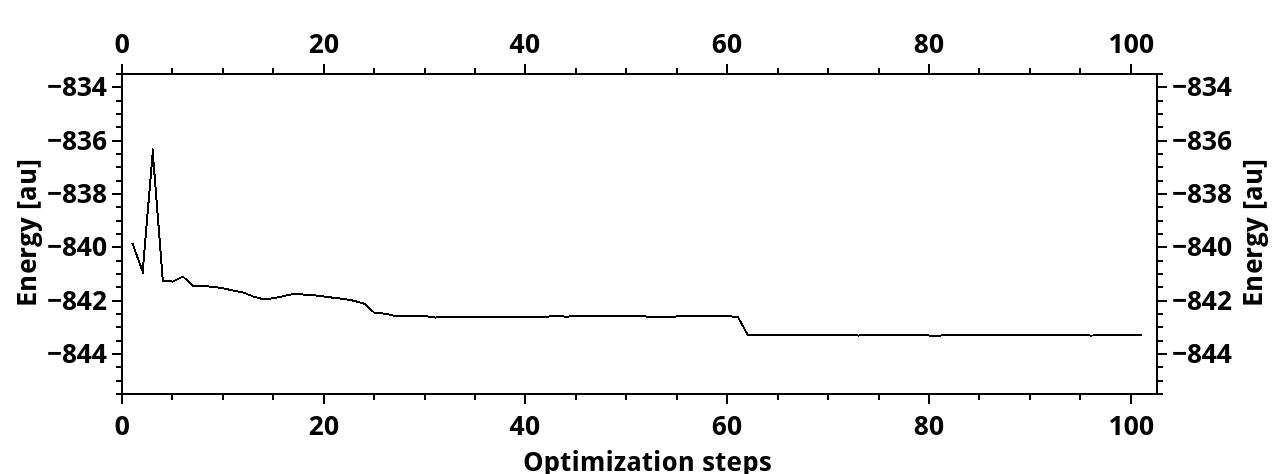
The same reduction of cutoffs that happened in benzene happens also for two graphene-based molecules. I made calculations also for a triangle of three benzene rings (13 carbon atoms+9 hydrogens) and of six benzene rings (22 carbon atoms+12 hydrogens). In both cases, the L_u reduces to about 3. Also, the optimization lasts much longer than it should, the energy was decreasing much longre than just few first varmin optimization steps.
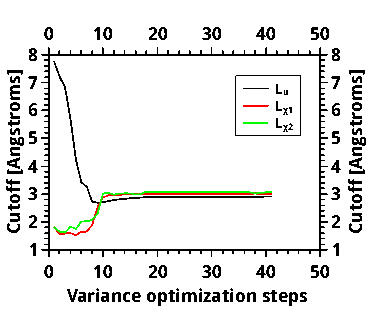
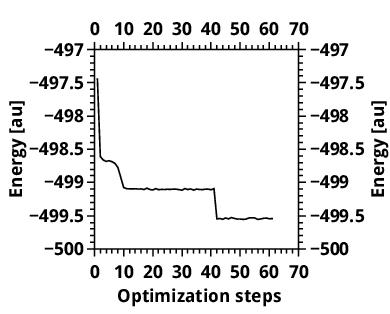
For the smaller molecule, I took the 40 variance optimization steps. The L_u cutoff reached a stable value and then, in the last steps, it slightly changed, suggesting that it did not reach its final value. I did not, at first, notice this final growth, and took some Jastrow factor from the “plateau”, optimized it with emin and fixed cutoffs, and ran DMC.
Here are the cutoff vs opt steps and energy vs opt steps (the latter contains also the energy-optimization run, that is the reason of the energy drop)
http://www.mediafire.com/convkey/e813/q ... 62mmfg.jpg
http://www.mediafire.com/convkey/7346/8 ... my8gfg.jpg
So the questions in this case are:
a) Is the DMC result reliable, or should I continue the variance optimization to get better cutoff, and then redo the DMC?
b) should I redo the VMC optimization starting from L_u closer to 3? Is there any way of determining the right initial value it this case? (should I for example run VMC optimization for fixed cutoffs for several L_u values and then look for smallest energy?)
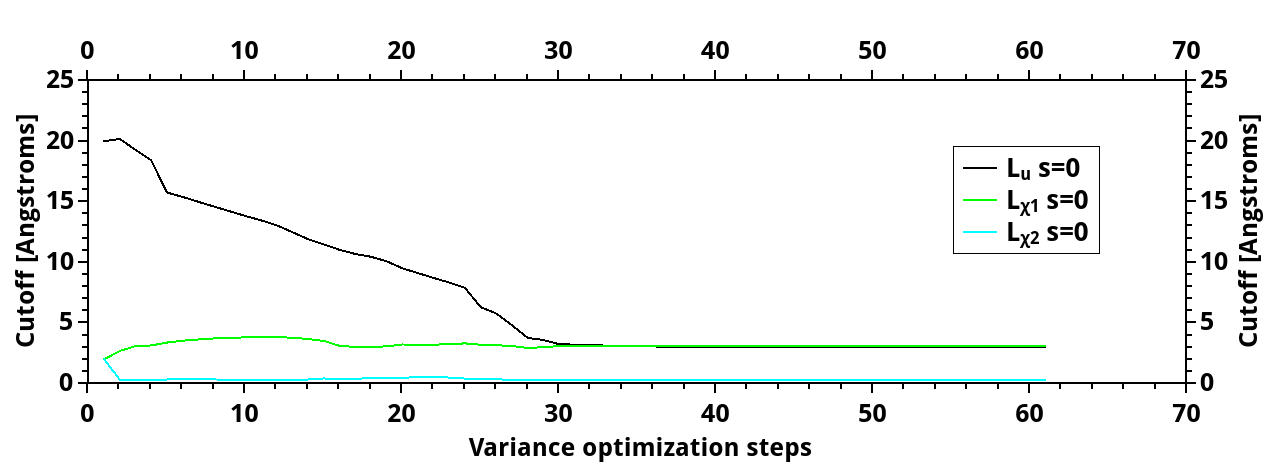
Then, for the 22-atom triangle, things are even more strange. I made only VMC (varmin with variable cutoffs + emin with fixed cutoffs), for a spin configuration with total spin s=0. Now, I have the same situation with L_u, and also, one of the L_chi drops significantly, to the value 0.30 while the bond length is about 1.42 or so. Does it indicate poor optimization quality?
The cutoff and energy evolution during optimization:
http://www.mediafire.com/convkey/7604/t ... 7pdffg.jpg
http://www.mediafire.com/convkey/8490/e ... u06xfg.jpg
BTW, is the energy rise in the beginning acceptable, or is it also a sign of poor optimization? should I increase vmc_nconfig_write?
Re: Cutoff lengths in the Jastrow factor
Posted: Mon Oct 14, 2013 4:40 pm
by Mike Towler
Hello,
Sorry to answer a long post with a short one (very busy today).
Two things:
(1) Large amounts of effort expended in getting the absolute best possible Jastrow factor are rarely reflected in improvements of subsequent DMC simulations.
(2) How many configs are you using?
M.
Re: Cutoff lengths in the Jastrow factor
Posted: Tue Oct 15, 2013 9:42 pm
by varelse
500 000 for both "graphene-based" molecules, 200 000 for benzene.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}